






La sindrome di Mowat Wilson (MWS) è una sindrome congenita rara, caratterizzata da anomalie multiple e da un particolare fenotipo (Garavelli,2007).
Fu descritta per la prima volta nel 1998 da due medici australiani, D.R. Mowat e M.J. Wilson e il gene responsabile della condizione, denominato ZEB2, è stato scoperto nel 2001 contemporaneamente da Cacheux in Francia e Wakamatsu in Giappone (Mowat,2003).
I soggetti si caratterizzano per un particolare fenotipo facciale, deficit cognitivo da moderato a grave, epilessia, e alcune malformazioni congenite tra cui malattia di Hirschsprung (HSCR), anomalie genito-urinarie ( in particolare ipospadia nei maschi), difetti cardiaci congeniti, agenesia del corpo calloso e anomalie degli occhi. Inoltre, diversi studi di analisi del genotipo e fenotipo dimostrano che la gestalt del viso e il ritardo psicomotorio sono caratteristiche costanti mentre le malformazioni congenite sono variabili.
In un piccolo numero di pazienti mutazioni inusuali possono portare a un fenotipo atipico (Garavelli,2007).
La prevalenza, ovvero il rapporto tra il numero di eventi sanitari rilevati in una popolazione in un definito momento e il numero degli individui della popolazione osservati nello stesso periodo, è ancora sconosciuta, ma sono stati descritti in tutto il mondo circa 200 casi clinici, di cui 30 in Italia. Tuttavia è probabile che questa incidenza sia sottostimata dal momento che la MWS non è sempre facilmente diagnosticabile, soprattutto se i pazienti non manifestano contemporaneamente la HSCR: diversi soggetti, infatti, possono non essere riconosciuti nell’infanzia e in alcuni casi fino all’età adulta (Mowat,1998).
La sindrome di Mowat Wilson è causata da un’alterazione genetica (delezione o mutazione) di un gene denominato Zinc finger homeobox 1 B (ZEB2), localizzato sul braccio lungo del cromosoma 2, in posizione 2q22. Queste alterazioni portano ad una perdita di funzione di una copia del gene (Mowat,1998).
L’alterazione genetica è quasi sempre sporadica ed il rischio di ricorrenza della patologia, cioè il rischio che si ripresenti in un altro figlio della stessa coppia di genitori, è intorno al 2%. Sono stati descritti rari casi di ricorrenza tra fratelli; questi eventi possono avvenire quando un genitore ha una piccola popolazione di cellule nell’ovaio o nel testicolo con la mutazione che causa la MWS e questo fenomeno è denominato mosaicismo germinale (McGaughran,2005).
Il gene ZEB2 (zinc finger E-box protein) fu identificato (Garavelli,2007) da Mowat e dai suoi colleghi che descrissero la sindrome nel 1998, identificando il locus del cromosoma 2q21-q23. Nel 2002, in seguito a vari studi, venne fatta un’associazione causale tra mutazioni del gene ZEB2 e MWS e nella letteratura internazionale furono identificati circa 200 casi (Garavelli,2007).
Più di 100 mutazioni di ZEB2 sono state identificate nelle persone affette dalla sindrome di Mowat Wilson. Queste mutazioni quasi sempre consistono in una inattivazione di una copia del gene, mentre, in alcuni casi, l’intero gene è cancellato e, in altri casi ancora, le mutazioni del gene portano ad una produzione eccessiva e non funzionale della proteina per cui il gene ZEB2 codifica (Mowat,1998).
La carenza di ZEB2 interferisce con la formazione di molti organi e tessuti del corpo umano prima della nascita. Un anormale sviluppo delle strutture derivate dalla cresta neurale, come il sistema nervoso e la gestalt facciale, sta alla base di molte caratteristiche della MWS. Il ruolo del gene ZEB2 nello sviluppo dei nervi che controllano l’apparato digerente potrebbe anche spiegare perché molte persone affette da MWS abbiano anche la HSCR, un disordine intestinale che causa una severa costipazione, blocco intestinale e allargamento del colon (Zweier at all,2005).
Nella sindrome di Mowat Wilson le caratteristiche del viso sono tipiche e su queste, infatti, si basa la diagnosi a cui di solito viene associato un grave ritardo mentale. Il fenotipo, tuttavia, cambia con l’età ed è per questo che risulta necessario fare riferimento alla diagnosi (Garavelli,2007).
Nell’infanzia si osserva un eccesso di pelle sulla nuca, il cranio ha la forma arrotondata, i capelli sono fini e radi, il collo è gonfio nella parte anteriore, il volto appare di forma quadrata e con fronte alta, con bozze frontali e ipertelorismo, gli occhi sono grandi ma infossati e possono presentare strabismo ed epicanto, il naso è caratterizzato da una punta arrotondata prominente, la bocca è spesso aperta e con il labbro superiore a forma di M. Inoltre, i soggetti appaiono spesso sorridenti e con il mento appuntito, stretto e dalla forma triangolare.
A queste caratteristiche si aggiunge il “telecanto”, ovvero un’aumentata distanza tra gli occhi con normale distanza tra le pupille e le orecchie ruotate posteriormente. Tuttavia le caratteristiche più frequenti e maggiormente riconoscibili sono le sopracciglia generalmente folte, ma rade nella parte centrale ed i caratteristici lobi delle orecchie, grandi e sollevati, caratterizzati da una depressione centrale.
Durante l'infanzia si osservano le seguenti caratteristiche: la faccia si allunga e il mento diventa più prominente, le sopracciglia sono spesso ampie e orizzontali, con una distanza maggiore l’una dall’altra e la columella diventa più evidente. Inoltre, i bambini comunemente hanno la bocca aperta e un’ espressione sorridente: la scialorrea, infatti, è una caratteristica significativa per alcuni di essi (Garavelli,2008).
Negli adolescenti e negli adulti, invece, la faccia si allunga con prognatismo e mento lungo, accompagnato anche dall’ allungamento della punta del naso (Mowat,1998).
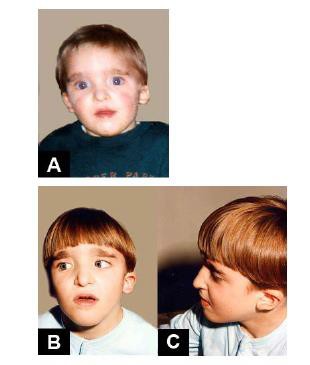

Figura 1: caratteristiche cliniche di bambini all’età di: (A) 1 anno e 6 mesi; (B-C) 5 anni; ( D-E) 13 anni e 8 mesi; (F-G) 18 anni. - Tratta da Garavelli,2007
I soggetti con MWS alla nascita hanno dei parametri di crescita che rientrano nella norma: il peso di un soggetto nato a termine è di media 3370g (25-50° percentile), la lunghezza media è di 50,9 cm ( 50- 75° centile),la circonferenza cranica è di 33 cm ( 3-10 centile), che risulta essere un percentile inferiore.
La microcefalia è a volte presente alla nascita ma, più spesso, si sviluppa gradualmente nell’infanzia e non necessariamente in tutti i bambini: è un segno comune ma non costante e, infatti, è presente in 135 di 166 soggetti dei casi pubblicati.
La maggior parte dei pazienti sono di corporatura esile (Garavelli,2007).
I bambini, soprattutto nei primi anni di vita, hanno un tono alterato manifestando ipotonia e ritardo delle principali tappe di sviluppo, ad esempio iniziano a stare seduti senza supporto solo all’età di 20 mesi, passando poi per la navigazione a crociera e raggiungendo il cammino all’età media di 4 anni. Anomalie sono riscontrate anche nell’ andatura, che si presenta a base allargata con le braccia spesso tenute flesse ai gomiti, e nell’articolazione del discorso. Quest’ultimo è assente o è limitato a poche parole pronunciate all’età di 5-6 anni (Mowat,1998).
Meno compromesse risultano, d’altra parte, le loro capacità ricettive e di comprensione: mostrano infatti di capire cosa viene loro detto piuttosto che esprimere i proprio bisogni e desideri.
Alcuni pazienti affetti da MWS sono in grado di servirsi della comunicazione aumentativa alternativa o del linguaggio dei segni o immagini o strumenti elettronici di comunicazione.
Dal punto di vista comportamentale i soggetti con MWS appaiono spesso felici e sorridenti ed estremamente socievoli e affettivi e ciò non può che rappresentare, sicuramente, un loro punto di forza.
Altre caratteristiche che riguardano il fenotipo comportamentale frequenti nelle persone affette da MWS sono: ricorso di atteggiamenti orali come digrignare i denti, mettere in bocca e masticare oggetti o parti del corpo, e scialorrea. Essi manifestano anche comportamenti stereotipati o ripetitivi quali colpire, battere, sfogliare le pagine. Sono stati riscontrati in diversi soggetti disturbi depressivi e dell’umore. I soggetti risultano particolarmente felici e sorridenti ma mostrano anche atteggiamenti di auto ferimento (Niemczyk, 2017).
La cardiopatia congenita nei casi affetti da MWS è stata dimostrata in 87 pazienti su 167 dei casi pubblicati (Garavelli,2007) ed è la conseguenza di un'alterazione del normale sviluppo embrionale del cuore nelle prime quattro-dieci settimane di gravidanza.
La malformazione cardiaca può essere di vario tipo: si va da anomalie che riguardano una singola parte del cuore (per es. una valvola) ad anomalie molto complesse in cui c'è un grave sovvertimento dell'intera architettura cardiaca.
L’ampio spettro dei difetti cardiaci include: pervietà del dotto arterioso (16 pazienti), stenosi polmonare (12 pazienti), difetto del setto interventricolari (12 pazienti), difetto del setto atriale (8 pazienti), sling dell’arteria polmonare (6 pazienti), Tetralogia di Fallot (5 pazienti), coartizione aortica (4 pazienti), atresia polmonare (1 pazienti), stenosi periferica dell’arteria polmonare (1 paziente), interruzione dell’arco aortico (1paziente), prolasso mitralico (1 paziente), stenosi della valvola aortica ( 1 paziente) (Garavelli,2007).
Nel 2007 lo studioso Dastot-LeMoal ha suggerito che lo sling dell'arteria polmonare, con o senza stenosi tracheale (anello vascolare anomalo determinato dall’origine anomala dell’arteria polmonare sinistra dalla parte destra del cuore) può avere una particolare associazione con la MWS (Garavelli,2007).
Il coinvolgimento neurologico è una caratteristica principale nei soggetti con MWS e quasi tutti i segni clinici associati possono essere spiegati da un difetto nell’induzione, migrazione e differenziazione delle cellule della cresta neuronale.
In tutti i pazienti è presente una disabilità intellettiva che va da moderata a grave (Garavelli,2016). Come è stato già precisato precedentemente, le principali tappe di sviluppo appaiono in ritardo: i bambini stanno seduti senza supporto solo all’età di 20 mesi, camminano intorno ai 4 anni, il linguaggio è limitato a poche parole (Garavell,2007).
Diversi studi indicano svariate anomalie cerebrali presenti nella sindrome di Mowat Wilson. Tra i più frequenti troviamo agenesia del corpo calloso (ACC) e ipoplasia del corpo calloso ( HCC).
Per quanto riguarda il ruolo di ZEB2 sull’anatomia macro strutturale del cervello è stato condotto, recentemente, da Garavelli et all uno studio su 54 pazienti (23 maschi e 31 femmine), i quali sono stati sottoposti a risonanza magnetica (MRI) con l’obiettivo di valutare nuove caratteristiche e aggiornare i pochi dati già presenti in letteratura. Dai suddetti studi è stato osservato che una caratteristica distintiva, presente in più della metà dei pazienti, è un difetto commisurale. Sono state riscontrate, infatti, anomalie del corpo calloso nella maggior parte dei pazienti con completa agenesia del corpo calloso (ACC), parziale ACC o ipoplasia con assottigliamento del corpo calloso; nel 40% dei pazienti si è osservato riduzione dello spessore della sostanza bianca e anomalie dei ventricoli cerebrali.
Risultati meno comuni ma comunque rilevanti sono: anomalie dei gangli della base, polimicrogiria, eterotopia periventricolare nodulare, displasia corticale, ipoplasia cerebellare, malformazioni Chiari di tipo 1 (Garavelli,2016).
Un’altra caratteristica tipica della sindrome è l’epilessia presente nel 70%- 75% dei casi (Cordelli,2013). L’insorgenza di crisi epilettiche, sempre di tipo focale, è stata osservata a un età media di 14,5 mesi. Questa ha inizio prima dell’età di 5 anni ma, raramente, nel primo anno di vita.
Non è emerso dagli studi condotti un tipo di crisi tipico della MWS ma sono state segnalate crisi generalizzate tonico-clonico, assenza, miocloniche e convulsioni focali (Cordelli,2013).
Oltre alle patologie sopra descritte, in molti pazienti si verificano anche anomalie muscolo-scheletriche. La maggior parte dei soggetti con MWS presenta una corporatura piuttosto snella, dita affusolate e sottili con punte prominenti. Altrettanto prominenti sono le articolazioni interfalangee che si sviluppano a partire dalla tarda infanzia. Anche nei piedi si registrano delle anomalie: questi, infatti, vengono descritti come piedi piatti con lieve calcagno valgo. Con quest’ultimo termine si intende un abbassamento della volta plantare, spesso con un conseguente valgismo del retropiede ed una abduzione dell’avampiede.
Altra caratteristica riscontrata sono le ossa “Wormian” ovvero piccole ossa formate all’interno da suture isolate dai centri di ossificazione tra i maggiori componenti della volta cranica.
Malformazioni sono state rilevate anche a livello dello sterno: il petto escavato ed il petto carenato. Il petto escavato è un’anomalia congenita della gabbia toracica, a livello del piano sterno-costale, che consiste in un’angolatura dello sterno verso l’interno, in direzione della colonna vertebrale. Questa malformazione non interessa però le coste, che, al contrario, risultano perfettamente conformate. Inoltre, genera, solitamente, dei problemi legati alla depressione del torace, che, oltre a generare un evidente problema estetico, può compromettere in modo sostanziale l’attività cardio-respiratoria.
Al contrario, il petto carenato è una protrusione anteriore dello sterno. Ad esso si associa anche la depressione laterale di una parte delle cartilagini costali, in conseguenza della quale si producono delle concavità nella parte destra e sinistra del torace.
Molti dei bambini affetti da MWS presentano anche la scoliosi che, essendo abbastanza frequente, può anche essere vista come conseguenza delle altre malformazioni muscolo-scheletriche già citate. Frequente è anche il ginocchio valgo, così come le lussazioni non traumatiche alla rotula, le lievi contratture dell’anca, dei gomiti e delle ginocchia, le deviazioni radiali dei pollici, i pollici addotti, la sindattilia del primo-secondo dito della mano, ovvero la fusione di due dita della mano, gli alluci lunghi grandi e valghi ed altre anomalie specifiche delle dita dei piedi (Garavelli,2007).
I soggetti con sindrome di Mowat Wilson presentano anche delle anomalie oculari.
In uno studio del 2014 condotto da A. Bourchany et all sono stati descritti quattro pazienti con malformazioni oculari che si sono aggiunti ai casi riportati nella letteratura precedente (Bourchany,2014).
In questi nuovi soggetti le anomalie sono state riscontrate allo stesso modo sia nei maschi sia nelle femmine. Le malformazioni interessano tutti i segmenti dell’occhio e si presentano sempre in modo asimmetrico (12/12 pazienti) o anche unilaterale (7/12 pazienti).
Le malformazioni portano spesso a compromettere gravemente le facoltà visive. Tra le più diffuse si annoverano la macroftalmia, ovvero la presenza di un occhio che, a fronte di una struttura normale, è di dimensioni ridotte rispetto all’altro, e con una capacità visiva inferiore; il coloboma di retina,dell’ iride o del disco ottico, ossia una malformazione che colpisce una delle suddette strutture dell’occhio causata da una mancata chiusura di una fessura dell’occhio nella fase intrauterina, durante il primo- secondo mese di gestazione. Le conseguenze di un coloboma posso essere lievi o severe in relazione alla localizzazione del difetto congenito e dalla sua estensione: se è limitato all’iride non presenta problemi alla capacità visiva, mentre se si estende anche al nervo ottico ed alla retina porta ad una visione deficitaria (Bourchany, 2014).
In un solo paziente affetto da MWS è stata riscontrata un’anomalia di Axenfeld (Garavelli,2007), ovvero un disordine autosomico dominante che interessa il segmento anteriore dell’occhio e le strutture non oculari. Segni evidenti di questa sono: l’ipoplasia dell’iride, l’ adesione tra lo stroma dell’iride e l’angolo irido- corneale e l’aumento della pressione intraoculare. Esso presentava, inoltre, caratteristiche non oculari come ipoplasia maxillare, ovvero un ridotto sviluppo delle ossa mascellari, spesso riscontrabile in sindromi e malattie congenite, micro e macro anodontia, cioè una rara malattia genetica caratterizzata dalla mancanza di tutti i denti decidui o permanenti, ipospadia nei maschi e pelle periombelicale ridondante.
Lo strabismo è, invece, raramente menzionato anche se si risulta evidente in alcune fotografie.
Un altro difetto rilevato è il nistagmo che, a causa di difficoltà di fissaggio, è spesso descritto nella prima infanzia ma tende a risolversi con l’età.
Alla luce di quanto è stato descritto è necessario, quindi, effettuare una visita oftalmologica in età pediatrica per tutti i soggetti affetti da MWS (Garavelli,2007).
Tra le anomalie sino ad ora elencate devono essere annoverate anche quelle che interessano l’apparato urogenitale e renale (51% dei casi).
Infatti, 63 maschi su 100 affetti da MWS, per quanto non sia possibile fornire stime definitive, presentano anomalie genitali, in particolare l’ipospadia, presente nel 52% dei pazienti presi in considerazione. Dalle ultime revisioni in letteratura risulta essere l’anomalia più frequente del tratto urogenitale (Garavelli,2007). Questa è una malformazione del pene dovuta alla mal posizione del meato uretrale lungo la faccia ventrale con conseguente curvatura ventrale del pene ed anomala distribuzione della cute con aspetto a cappuccio.
Altre anomalie presenti sono criptorchidismo, lo scroto bifido, il reflusso vescico ureterale, ossia il reflusso di urina dalla vescica all’uretere ed alla pelvi renale e l’idronefrosi, cioè la dilatazione del pelvi renale e dei calici renali dovuta ad una anomalia della giunzione tra il rene e l’uretere, che comporta un ostacolo al passaggio delle urine dalle cavità renali all’uretere e quindi alla vescica (Amiel,2008).
Un’altra malattia che caratterizza i soggetti con sindrome di Mowat Wilson è il morbo di Hirschsprung (HSCR), ovvero la congenita assenza di cellule gangliari enteriche nella parte distale del colon (Coyle,2015).
Questo può presentarsi in due forme differenti:
In un recente studio condotto da Coyle e Puri nel 2015, incorporando i dati di 256 pazienti con MWS, si è visto che HSCR è diagnosticata in 111 pazienti corrispondente al 43% dei casi.
Pur non essendo sempre presente, come evidenzia lo studio di Coyle, e pur essendo caratteristiche di altre sindromi, come per esempio la trisomia 21 o la sindrome di Goldberg-Shprintzen, malattia estremamente rara (finora sono stati descritti meno di 50 casi) che è di solito caratterizzata da un habitus Marfanoide associato a anomalie craniofacciali, scheletriche e cardiovascolari, e difficoltà di apprendimento, rimane comunque uno dei criteri diagnostici per la sindrome di Mowat Wilson (Garavelli,2007).
Un altro elemento problematico riscontrato nei bambini affetti da Sindrome di Mowat Wilson e su cui si stanno conducendo degli studi negli ultimi anni è il disturbo del sonno.
In uno studio condotto da Evans et all (2015) si è indagato sul disturbo del sonno che è stato riportato dalla maggior parte delle famiglie coinvolte con interviste non strutturate, le quali riscontrano frequente veglia durante la notte e risveglio nelle prime ore dell’alba. Questo studio ha coinvolto 35 soggetti con MWS, ai quali è stato somministrato sia la Scala del disturbo del sonno per bambini (SDSC) sia la Behaviour Checklist Developmental (DBC) per misurare i disturbi comportamentali ed emotivi. Dai risultati emerge che è comune un disturbo del sonno nella maggior parte dei soggetti con sindrome di Mowat Wilson sia nella fase di veglia, sia per quanto riguarda la respirazione che si ha durante il sonno che la sonnolenza diurna. Inoltre è dimostrata una correlazione tra i disturbi del sonno e i disturbi di comportamento.
Un recente studio condotto da Niemczyk et all nel 2016 evidenzia un altro disturbo che può trovarsi in associazione con la sindrome di Mowat Wilson ovvero l’incontinenza e altri sintomi fisiologici. L’incontinenza funzionale (compresa enuresi notturna, incontinenza urinaria giornaliera e incontinenza fecale) è un disturbo comune che colpisce l’1-10% dei soggetti con MWS e solitamente diminuisce con l’avanzare dell’età. Tuttavia nei soggetti con grave disabilità intellettiva (ID) la percentuale rimane più alta anche con l’avanzare dell’età. (Niemczyk et all, 2016)
La sindrome di Mowat Wilson di solito è diagnosticata durante l’infanzia attraverso un’approfondita valutazione clinica, l’identificazione delle caratteristiche fisiche e del viso e le informazioni rilevate da una varietà di test specializzati. Per esempio si effettua un’analisi citogenetica per escludere delezioni o traslocazioni; l’analisi FISH rileva eliminazioni submicroscopiche; il sequenziamento della sequenza codificante completa di ZEB2 identifica la mutazione; la fluorescenza semiquantitativa a reazione multiplex a catena della polimerasi (PCR) permette di individuare altri riarrangiamenti sfuggiti ai metodi convenzionali.
Particolarmente importante nel diagnosticare tale sindrome risulta essere la gestalt facciale. Gravi malformazioni come HSCR, malattie cardiache, agenesia del corpo calloso sono comuni anche se non sempre presenti. Inoltre, il ritardo delle tappe dello sviluppo è sempre costante.
Solo un piccolo numero di pazienti con mutazioni rare può mostrare un quadro atipico.
E’ necessario per la diagnosi sottoporre ad analisi molecolare il gene ZEB2.
Pur essendo i tratti del viso caratteristici, la presenza di HRCR epilessia e ritardo mentale possono far pensare si tratti della sindrome di Goldberg-Shprintzen (GOSHS) per questo è necessaria la diagnosi differenziale.
I soggetti con MWS sono spesso descritti come aventi un’andatura a base allargata o atassica, spesso sorridenti e felici e con una personalità socievole. Queste caratteristiche sono comuni con la sindrome di Angelman pur rimanendo diverse le caratteristiche del viso (Garavelli,2007).































